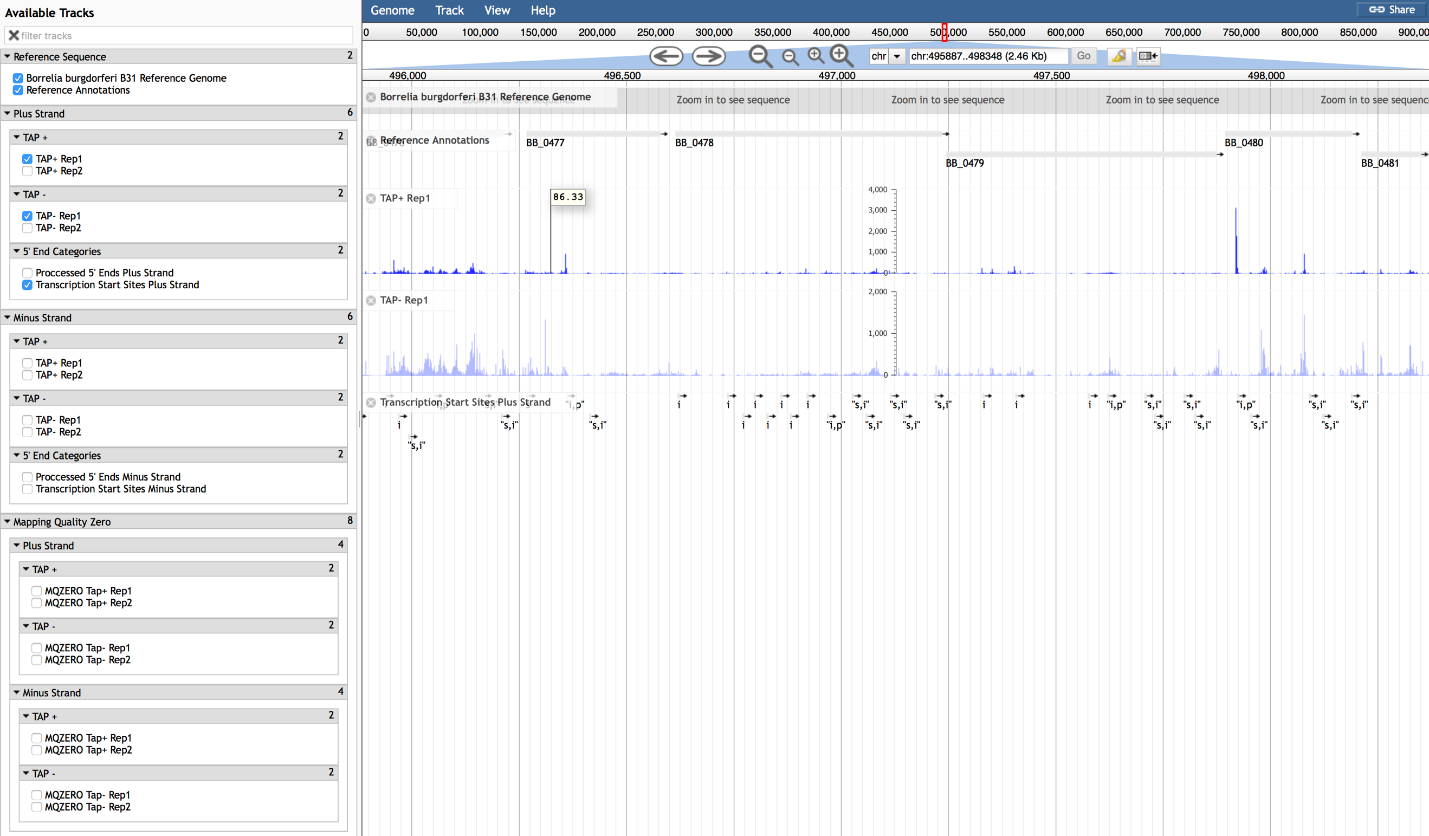
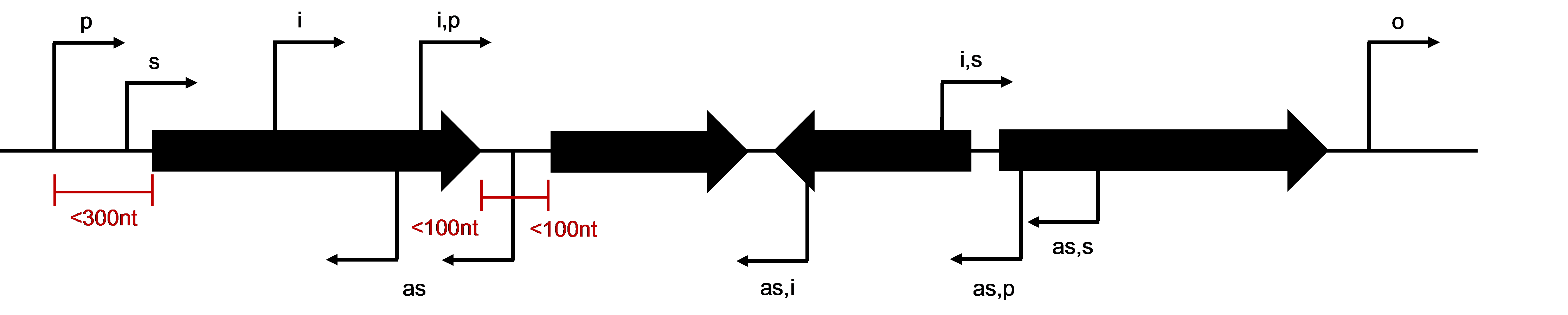This page contains a link to the B. burgdorferi 5’ end transcriptome and instructions for navigating the dataset. Transcriptional start sites (TSSs) and processed 5’ ends were distinguished with differential application of tobacco acid pyrophosphatase (TAP) in the RNA-seq library preparations. These data will allow users to accurately determine gene boundaries, identify putative leaderless and long untranslated regions, and discover novel transcripts. For specific examples, experimental validation of the data and more information on global analysis of the B. burgdorferi 5’ end transcriptome please refer to our complete publication.
Data are displayed using JBrowse, developed by Skinner at al., 2009. (Skinner at al., 2009. JBrowse: A next-generation genome browser. Genome Research. 19:1630-1638 doi:10.1101/gr.094607.109). More information concerning JBrowse can be found here: http://jbrowse.org